Nitrilek
A nitrilek olyan szerves vegyületek, amelyek −C≡N funkciós csoportot tartalmaznak.[1] A −CN csoportban a szénatom és a nitrogénatom között hármas kötés található. A szerves vegyületek nevezéktana szerint „-(karbo)nitril” végződést kapnak, ha a nitril a legnagyobb prioritású csoport (például akrilnitril), ha nem ez a fő funkció, akkor pedig „ciano-” előtagot (például 2-ciano-2-metilpropánsav).

A -C≡N csoportot tartalmazó szervetlen vegyületeket nem nitrileknek, hanem cianidoknak nevezik.[2]
Habár mind a nitrilek, mind a cianidok cianidsókból származtathatóak, a nitrilek túlnyomó többsége sokkal kevésbé mérgező, mint a cianidok. Sok hasznos vegyületben találhatók nitrilek, a metil-cianoakrilátot például a pillanatragasztókban és a butadién-nitril kaucsukban használják, mely többek között a latexmentes laboratóriumi és orvosi gumikesztyűk anyaga.
Szerkezetük és tulajdonságaik
szerkesztésA nitrilekben található N−C−C váz lineáris, ami a hármas kötésű szénatom sp hibridizációját tükrözi. A C−N kötéstávolság – a hármas kötéssel összhangban – rövid, 116 pm.[3] A nitrilek poláris vegyületek, dipólusmomentumuk nagy. Folyadék halmazállapotban dielektromos állandójuk nagy, nem ritkán 30 körüli érték.
Felfedezésük
szerkesztésA nitrilek homológ sorának első tagját, a hangyasav nitriljét, a hidrogén-cianidot elsőként C.W. Scheele szintetizálta 1782-ben.[4][5] 1811-ben J. L. Gay-Lussac-nak sikerült tiszta formában előállítania a nagyon mérgező és illékony savat. A benzoesavak nitriljét elsőként Friedrich Wöhler és Justus von Liebig állították elő, de a szintézis alacsony hozama miatt nem tudták meghatározni fizikai és kémiai tulajdonságaikat, és szerkezetükre sem tudtak javaslatot tenni. 1834-ben Théophile-Jules Pelouze propionitrilt szintetizált, és úgy gondolta, hogy az a propil-alkohol és a hidrogén-cianid észtere.[6] A benzonitril Hermann Fehling által 1844-ben kidolgozott – ammónium-benzoát hevítésével történő – szintézise volt az első módszer, mellyel a kémiai kutatáshoz elegendő mennyiségű anyagot sikerült előállítani. Fehling a vegyület szerkezetét úgy határozta meg, hogy saját eredményeit a hidrogén-cianid ammónium-formiát hevítésével végzett szintézisével hasonlította össze. Az újonnan létrehozott anyagnak ő adta a nitril nevet, mely aztán ennek a vegyületcsoportnak a nevévé is vált.[7]
Szintézisük
szerkesztésA nitrilek ipari előállításának két fő módja az oxidatív ammonolízis (ammoxidáció) és a „hidrocianálás”. Mindkét módszer a zöld kémia tárgykörébe illik abban az értelemben, hogy az eljárások során nem keletkezik sztöchiometrikus mennyiségű só. Nitrileket számos más módszerrel is elő lehet állítani, ezeket általában különlegesebb alkalmazások esetén használják.
Ammoxidáció
szerkesztésAz ammoxidáció során a szénhidrogént részlegesen oxidálják ammónia jelenlétében. Ezt az eljárást ipari léptékben végzik az akrilnitril előállításához:[8]
- CH3CH=CH2 + 3/2 O2 + NH3 → NCCH=CH2 + 3 H2O
Az akrilnitril mellett melléktermékként acetonitril keletkezik. A benzonitril, ftalonitril, valamint az izobutironitril legtöbb származékát is ammoxidációval állítják elő. Az eljárás során katalizátorként fém-oxidokat használnak, a reakció feltehetően aldehid köztiterméken keresztül játszódik le.
Hidrocianálás
szerkesztésA hidrocianálás gyakori módszer a nitrileknek alkénekből és hidrogén-cianidból történő előállítására. A folyamat homogén katalízissel valósítható meg. A hidrocianálás egyik példája az adiponitril előállítása buta-1,3-diénből kiindulva:
- CH2=CH-CH=CH2 + 2 HCN → NC(CH2)4CN
Ciánhidrinek
szerkesztésA ciánhidrinek a nitrilek különleges csoportja, melyek cianidion aldehidekre történő addíciója során (ciánhidrin-reakció) keletkeznek. A szerves karbonilcsoport polaritása miatt – az alkének hidrocianálásától eltérően – a reakció nem igényel katalizátort.
Szerves halogénvegyületekből és cianidsókból
szerkesztésAlifás nitrileket Kolbe-féle nitrilszintézissel alkil-halogenidekből fém-cianidokkal végzett nukleofil szubsztitúció révén állítanak elő. Aril-nitrilek aril-halogenidből réz(I)-cianid segítségével magas hőmérsékleten állíthatók elő, ez a Rosenmund–von Braun-szintézis.
Amidok és oximok dehidratálása
szerkesztésNitrilek előállíthatók primer amidok dehidratálásával. Erre a célra számos reagens használható, ezek egyike a benzamid benzonitrillé történő alakításához használt etil-diklórfoszfát és DBU kombinációja:[9]

- Ennek a reakciónak két köztiterméke az A amid tautomer és ennek B foszfát adduktuma.

A szekunder amidok dehidratálása (von Braun-féle amid lebontás) is nitrilt eredményez, e reakció során az egyik C-N kötés felhasad. Az aldoximok (RCH=NOH) dehidratációja során is nitrilek keletkeznek. Ennek az átalakulásnak a jellemző reagensei a trietil-amin/kén-dioxid, zeolitok vagy szulfuril-klorid. Ennek az eljárásnak az egyik felhasználása a nitrilek aldehidekből, hidroxilaminból és nátrium-szulfátból történő one-pot szintézise.[10]

Sandmeyer-reakció
szerkesztésAromás nitrileket laboratóriumban gyakran anilinből kiindulva állítanak elő diazóniumsón keresztül, ezt nevezik Sandmeyer-reakciónak. A reakcióhoz átmenetifém-cianid szükséges.[11]
- ArN2+ + Cu2CN2 → ArCN + N2 + 2 Cu+
Egyéb eljárások
szerkesztés- A cianocsoport egyik kereskedelmileg elérhető forrása a dietilalumínum-cianid (Et2AlCN), mely trietilalumíniumból és HCN-ból állítható elő.[12] Ketonokra történő nukleofil addíciós reakciókban használták.[13]
- A cianidionok elősegítik a dibromidok gyűrűzárását. α,α'-dibróm adipinsav nátrium-cianiddal etanolban végzett reakciója során ciano-ciklobután származék keletkezik:[14]

- Az úgynevezett Franchimont-reakció (A. P. N. Franchimont, 1872) során α-brómkarbonsav dimerizálódik a cianocsoport hidrolízisét és dekarboxileződést követően[15]

Reakcióik
szerkesztésA szerves nitrilek – a reaktánsoktól és a körülményektől függően – sokféle reakcióban vehetnek részt. A cianocsoport hidrolizálható, redukálható, vagy cianidionként távozhat a molekulából.
Hidrolízis
szerkesztésAz RCN nitrilek savas vagy bázikus körülmények között végzett hidrolízise külön lépésekben megy végbe, melynek eredménye először RC(=O)NH2 karboxamid, majd RCOOH karbonsav. A nitrilek hidrolízisét általában a karbonsavak előállításának egyik legjobb módszerének tekintik, azonban ezen sav vagy bázis által katalizált reakcióknak vannak bizonyos korlátai és/vagy hátrányai az amidok előállításában. Általános korlát, hogy a sav vagy bázis utolsó lépésben történő semlegesítése jelentős sóképződéssel jár, ami szennyezi a terméket. Konkrét példán bemutatva:
- Bázis katalizált reakciók. Kinetikai vizsgálatok alapján megbecsülhető a reakció egyes hidratációs lépéseinek relatív sebessége, és egy jellemző példaként az acetonitril és acetamid hidroxidion által katalizált hidrolízisének másodrendű sebességi állandója rendre 1,6·10–6, illetve 7,4·10– M−1s−1. A két érték összehasonlításából látható, hogy a bázis katalizált reakció második lépése gyorsabb, mint az első lépés, így a reakció nem áll meg az amid képződésénél, hanem továbbmegy a végtermékig (a karbonsav sóig). Ez azzal jár, hogy a hagyományos fémmentes bázis katalizált reakcióval előállított amid karbonsavakkal szennyezett lesz, és csak mérsékelt kitermelés érhető el.
- Sav katalizált reakciók. Erősan savas oldatok alkalmazásakor gondosan ügyelni kell a hőmérséklet és a reagensek arányának szabályozására, hogy – a hidrolízis exoterm jellege által is elősegített – polimerképződést elkerüljék.[18]
Redukció
szerkesztésA nitrilek nikkel katalizátor mellett hidrogénnel redukálhatók, a reakció során amin keletkezik (lásd nitril redukció). A Stephen-redukció során a nitril iminné redukálódik, mely hidrolízis során aldehiddé alakul.
Alkilezés
szerkesztésAz alkilnitrilek kellően savasak ahhoz, hogy karbaniont tudjanak képezni, mely számos elektrofil alkilezésére képes. E kivételes nukleofilicitásban kulcsfontosságú a -CN egység kis térigénye és negatív induktív effektusa. Mindezen sajátosságok miatt a gyógyszerkémiai szintézisekben a nitrilek ideálisak új szén-szén kötés kialakítására sztérikusan zsúfolt környezetben. Az újabb eredmények szerint a fém ellenion természetétől függően különböző koordináció alakul ki a nitril nitrogénjéhez vagy a szomszédos nukleofil szénatomhoz, ami gyakran alapvető különbséget okoz a reaktivitásban és sztereokémiában.[19]
Nukleofilek
szerkesztésA nitrilek szénatomja nukleofil addíciós reakciókban elektrofilként viselkedik:
- szerves cinkvegyületekkel szemben a Blaise-reakcióban
- alkoholokkal szemben a Pinner-reakcióban
- a szarkozin nevű amin ciánamiddal végzett reakciója során kreatin keletkezik[20]
- Houben–Hoesch-reakció során a nitrilek Friedel–Crafts-acilezéssel ketonokká alakulnak
További reakciók és vegyületek
szerkesztés- A reduktív deciánozás során a nitrilcsoport helyére proton lép.[21] Az egyik hatékony deciánozási módszer a hexametilfoszforamiddal (HMPA) és fémkáliummal terc-butanolban végrehajtott Birch-redukció. α-amino-nitrilek lítium-alumínium-hidriddel deciánozhatók.
- bázis jelenlétében nukleofil addícióval önmagukkal reagálnak a Thorpe-reakcióban
- Grignard-vegyülettel ketonná alakíthatóak
- A fémorganikus kémiában a nitrilekről ismert, hogy a karbociánozás során alkinekre addícionálódnak:[22]

A nitrilek származékai
szerkesztésSzerves ciánamidok
szerkesztésA ciánamidok R1R2N−CN általános szerkezetű N-ciano vegyületek, melyek a szervetlen ciánamid származékainak is tekinthetők.
Nitril-oxidok
szerkesztésA nitril-oxidok általános szerkezete R−CNO.

Előfordulásuk és felhasználásuk
szerkesztésA természetben a nitrilek számos növény- és állatfajban megtalálhatók. Több mint 120 természetes nitrilt izoláltak szárazföldi és tengeri forrásokból. A nitrilek gyakran megtalálhatók a csonthéjas magokban, különösen a mandulákban, valamint a megfőzött káposztafélékben (pl. káposzta, kelbimbó, karfiol), melyekben hidrolízis révén keletkezik. A ciánhidrinek közé tartozó mandelonitril a mandula és néhány más csonthéjas elfogyasztása révén keletkezik, és belőle hidrogén-cianid szabadul fel, ami a cianogén glikozidok okozta mérgezés felelőse.
Jelenleg 30-nál is több nitrilcsoportot tartalmazó gyógyszerhatóanyag van forgalomban, nagyon különféle orvosi javallatokkal. Ezeken kívül további legalább 20 nitrilcsoportot tartalmazó gyógyszerhatóanyag-jelölt áll klinikai fejlesztés alatt. A nitrilcsoport eléggé robusztus, a legtöbb esetben nem metabolizálódik, hanem változatlanul jut át a szervezeten. A nitrilcsoportot tartalmazó gyógyszerek nagyon sokfélék, a cukorbetegség elleni vildagliptintől az anasztrazolig, a mellrák elleni fő hatóanyagig terjed a skála. Egyes esetekben a gyógyszermolekula nitrilcsoportja révén egy enzim szubsztrát funkcionalitását utánozza, de a nitrilcsoport növelheti a vízoldékonyságot, vagy csökkentheti a máj oxidatív metabolizációjával szembeni érzékenységet.[23]
A nitrilcsoport számos gyógyszerben megtalálható.
-
 A periciazin nevű antipszichotikumot az ópiátfüggőség kezelésére vizsgálják
A periciazin nevű antipszichotikumot az ópiátfüggőség kezelésére vizsgálják -
 A szelektív szerotonin visszavétel gátlók közé tartozó antidepresszáns hatóanyag, a citaloprám szerkezete
A szelektív szerotonin visszavétel gátlók közé tartozó antidepresszáns hatóanyag, a citaloprám szerkezete -
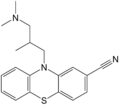 A ciamemazin nevű antipszichotikum
A ciamemazin nevű antipszichotikum -
 A mellrák kezelésére használt aromatáz inhibitor, a fadrozol szerkezete
A mellrák kezelésére használt aromatáz inhibitor, a fadrozol szerkezete -
 A szájon át szedhető, nem szteroid aromatáz inhibitor letrozolt a mellrák egyes fajtáinak kezelésére használják
A szájon át szedhető, nem szteroid aromatáz inhibitor letrozolt a mellrák egyes fajtáinak kezelésére használják





Jegyzetek
szerkesztés- ↑ IUPAC Gold Book nitriles
- ↑ NCBI-MeSH Nitriles
- ↑ Karakida, Ken-ichi, Tsutomu Fukuyama, and Kozo Kuchitsu. "Molecular Structures of Hydrogen Cyanide and Acetonitrile as Studied by Gas Electron Diffraction" Bulletin of the Chemical Society of Japan 1974, vol. 47, pp. 299-304.
- ↑ Lásd: * Carl W. Scheele (1782) "Försök, beträffande det färgande ämnet uti Berlinerblå" (Kísérlet a berlini kék színanyagával), Kungliga Svenska Vetenskapsakademiens handlingar (A Svéd Királyi Tudományos Akadémia Közleményei), 3: 264–275 (svéd nyelven).
- Latin kiadás: "De materia tingente caerulei berolinensis" in: Carl Wilhelm Scheele with Ernst Benjamin Gottlieb Hebenstreit (ed.) and Gottfried Heinrich Schäfer (trans.), Opuscula Chemica et Physica (Leipzig ("Lipsiae"), (Germany): Johann Godfried Müller, 1789), vol. 2, pages 148–174.
- ↑ David T. Mowry (1948). „The Preparation of Nitriles”. Chemical Reviews 42 (2), 189–283. o. DOI:10.1021/cr60132a001.
- ↑ J. Pelouze (1834). „Notiz über einen neuen Cyanäther”. Annalen der Pharmacie 10 (3), 249. o. DOI:10.1002/jlac.18340100302.
- ↑ Hermann Fehling (1844). „Ueber die Zersetzung des benzoësauren Ammoniaks durch die Wärme (Az ammónium-benzoát hőbomlása)”. Annalen der Chemie und Pharmacie 49 (1), 91–97. o. DOI:10.1002/jlac.18440490106. A 96. oldalon Fehling ezt írja: „Da Laurent den von ihm entdeckten Körper schon Nitrobenzoyl genannt hat, auch schon ein Azobenzoyl existirt, so könnte man den aus benzoësaurem Ammoniak entstehenden Körper vielleicht Benzonitril nennen.” (Mivel Laurent az általa felfedezett anyagot „nitrobenzoil”-nak nevezte – és már az „azobenzoil” is létezik –, ezért az ammónium-benzoátból származó anyagot talán „benzonitril”-nek nevezhetnénk.)
- ↑ Peter Pollak, Gérard Romeder, Ferdinand Hagedorn, Heinz-Peter Gelbke "Nitriles" Ullmann's Encyclopedia of Industrial Chemistry 2002, Wiley-VCH, Weinheim. doi:10.1002/14356007.a17_363
- ↑ Chun-Wei Kuo, Jia-Liang Zhu, Jen-Dar Wu, Cheng-Ming Chu, Ching-Fa Yao and Kak-Shan Shia (2007). „A convenient new procedure for converting primary amides into nitriles”. Chem. Commun. 2007 (3), 301–303. o. DOI:10.1039/b614061k. PMID 17299646.
- ↑ Sharwan K, Dewan, Ravinder Singh, and Anil Kumar (2006). „One pot synthesis of nitriles from aldehydes and hydroxylamine hydrochloride using sodium sulfate (anhyd) and sodium bicarbonate in dry media under microwave irradiation” (open access). Arkivoc, (ii) 41–44. o. [2007. szeptember 26-i dátummal az eredetiből archiválva]. (Hozzáférés: 2010. június 10.)
- ↑ o-Tolunitrile and p-Tolunitrile" H. T. Clarke and R. R. Read Org. Synth. 1941, Coll. Vol. 1, 514.
- ↑ W. Nagata and M. Yoshioka (1988). „Diethylaluminum cyanide”. Org. Synth.. ; Coll. Vol. 6: 436
- ↑ W. Nagata, M. Yoshioka, and M. Murakami (1988). „PREPARATION OF CYANO COMPOUNDS USING ALKYLALUMINUM INTERMEDIATES: 1-CYANO-6-METHOXY-3,4-DIHYDRONAPHTHALENE”. Org. Synth.. ; Coll. Vol. 6: 307
- ↑ Reynold C. Fuson, Oscar R. Kreimeier, and Gilbert L. Nimmo (1930). „Ring Closures In The Cyclobutane Series. Ii. Cyclization Of Α,Α′-Dibromo-Adipic Esters”. J. Am. Chem. Soc. 52 (10), 4074–4076. o. DOI:10.1021/ja01373a046.
- ↑ Franchimont Reaction
- ↑ J. Houben, Walter Fischer (1930) "Über eine neue Methode zur Darstellung cyclischer Nitrile durch katalytischen Abbau (I. Mitteil.)," Berichte der deutschen chemischen Gesellschaft (A and B Series) 63 (9): 2464 – 2472. doi:10.1002/cber.19300630920
- ↑ http://www.drugfuture.com/OrganicNameReactions/ONR197.htm Merck & Co., Inc., Whitehouse Station
- ↑ V. Yu. Kukushkin, A. J. L. Pombeiro, Metal-mediated and metal-catalyzed hydrolysis of nitriles (a review), Inorg. Chim. Acta, 358 (2005) 1–21
- ↑ Tetrahedron Volume 61, Issue 4, 24 January 2005, Pages 747–789 doi:10.1016/j.tet.2004.11.012
- ↑ Smith, Andri L.; Tan, Paula (2006). „Creatine Synthesis: An Undergraduate Organic Chemistry Laboratory Experiment”. J. Chem. Educ. 83, 1654. o. [2008. július 4-i dátummal az eredetiből archiválva]. DOI:10.1021/ed083p1654. (Hozzáférés: 2010. június 10.)
- ↑ The reductive decyanation reaction: chemical methods and synthetic applications Jean-Marc Mattalia, Caroline Marchi-Delapierre, Hassan Hazimeh, and Michel Chanon Arkivoc (AL-1755FR) pp 90–118 2006 Article[halott link]
- ↑ Yoshiaki Nakao, Akira Yada, Shiro Ebata, and Tamejiro Hiyama (2007). „A Dramatic Effect of Lewis-Acid Catalysts on Nickel-Catalyzed Carbocyanation of Alkynes” (Communication). J. Am. Chem. Soc. 129 (9), 2428–2429. o. DOI:10.1021/ja067364x. PMID 17295484.
- ↑ (2010. November) „Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore”. J Med Chem 53 (22), 7902–17. o. DOI:10.1021/jm100762r. PMID 20804202.
Fordítás
szerkesztésEz a szócikk részben vagy egészben a Nitrile című angol Wikipédia-szócikk ezen változatának fordításán alapul. Az eredeti cikk szerkesztőit annak laptörténete sorolja fel. Ez a jelzés csupán a megfogalmazás eredetét és a szerzői jogokat jelzi, nem szolgál a cikkben szereplő információk forrásmegjelöléseként.